




Have you ever wondered how scientists and bioinformaticians store, access, and organize the genetic variants they discover in each sample? This is the purpose of the Variant Call Format (VCF).
To tie in with what we've already discussed in previous posts, let's remember:
FASTQ files: We've learned what FASTQ files are and how to assess the quality of their sequencing data. These files store raw sequencing reads along with their corresponding quality scores.
FASTA files: These files are straightforward. They're used to store sequences, such as the reference genome sequence, without any additional information or annotations.
Alignment and BAM files: When the sequences in your FASTQ files are aligned to a reference sequence (usually stored in a FASTA file), the resulting alignments are typically represented in a BAM file.
Just as with the BAM file, the VCF file format has its own comprehensive specification document, which you can find here. In this post, I'll introduce the main features of the VCF file format.
Before we proceed...
What are variants?
In short, a variant refers to any definitive change in the DNA sequence compared to a reference genome.
For a more detailed explanation, let's delve into the context of human genomics, a field with which I am particularly familiar. This will provide additional information, just in case the brief definition wasn't sufficient.
The Human Genome Project was an international scientific research project that aimed to map and understand all the genes of human beings. The project formally began on October 1, 1990, and was declared complete in April 2003. Therefore, the duration of the Human Genome Project was approximately 13 years. If you want to know more about the project, wondering who gave blood to be sequenced, how much it cost and more access the NIH Fact Sheet website.
The outcome of the Human Genome Project was a reference genome, which is essentially a 'standard' or 'baseline' for human DNA. However, every individual has small differences in their DNA sequence compared to this reference. These differences are what we call variants.
Variants come in different forms:
Single nucleotide polymorphisms (SNVs): These are the most common type of genetic variation among people. Each SNV represents a difference in a single DNA building block, called a nucleotide. It's commonly called SNPs, but in this case, it is limited to germline DNA and must be present in at least 1% of the population.
Insertions and deletions (indels): These are small additions or losses of nucleotide bases in the DNA sequence.
Structural variants: These include larger-scale changes like inversions, where a section of DNA is flipped around, or copy number variations, where a segment of the DNA is duplicated or deleted.
The amount of data that arises from variant discovery in genomic data is enormous. Therefore, an efficient, compact, and flexible format was needed to store and share this information, which led to the creation of the VCF format.
Just a little of personal experience to share: after starting work on a feature in our system to provide raw data to clients I discovered that VCF is also a file extension for vCard. If you upload your VCF file to Google Cloud Storage it will automatically identify it as a text/vcard. The consequence is that if you generate a authenticated URL to make it available for download the user will complain about their system trying to open the variants in a contact manager program - and the program telling them that the file is corrupted.
Anatomy of a VCF file
A VCF file consists of meta-information lines, a header line, and then data lines each containing information about a position in the genome. The format also can contain genotype information on samples for each position.
The meta-information section (lines starting with '##') provides key-value pairs describing the version, reference and other important characteristics of the file. The header line (starting with '#') is a tab-separated list describing the data columns in the file. The following data lines contain information about specific variants.
Working with VCF files can involve various operations, including filtering variants based on certain criteria, annotating variants with additional information, comparing variants between different individuals or groups, and so on. There are many software tools available for working with VCF files, such as BCFtools, BEDtools, and the Genome Analysis Toolkit (GATK), among others.
The image below, extracted from the file format specification, shows the main sections.
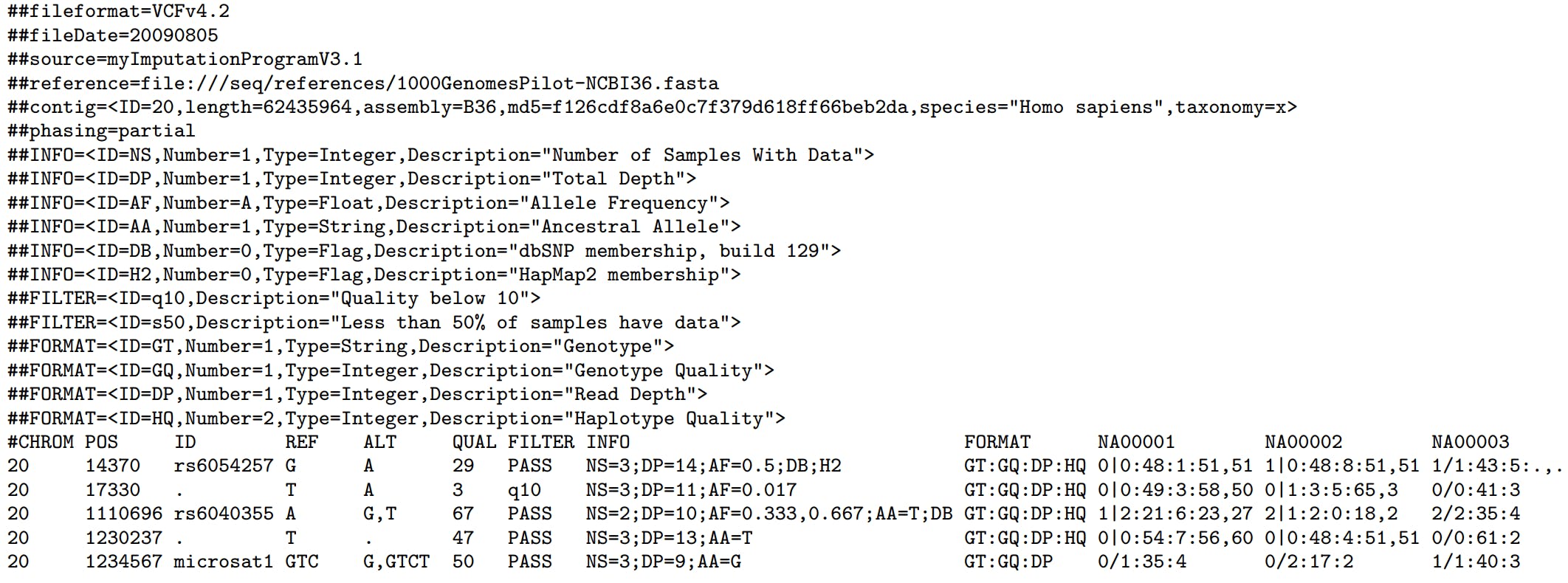
In this example, we see the header with meta-informations. For example, we know that these variants were identified using chromosome 20 of the reference genome b36 of length 62.435.964 pairs of base (pb). We know it because of the line starting with "##contig=". If not sure, we can check if it matches the reference we are using by comparing de md5 hash.
If you are confused about the different versions and nomenclatures of the human genome I recommend checking these two resources:
Human genome reference builds - GRCh38 or hg38 - b37 - hg19. From Broad Institute.
Which human reference genome to use? Text from Heng Li's Blog. We already mentioned him in the post about compleasm and spider genomes.
Also, as observed in the VCF image, there are five positions. For those of you who have experience working with intervals and VCF files, it's important to note: positions in VCF files are 1-based. You can find more about it in section 1.4.1 of the file pattern specification.
Each position has its ID. These ones starting with 'rs' came from dbSNP and you can learn more about it on this page from 23AndMe. Remember, a rsID does not uniquely define a variant. Instead, it refers to a particular genomic position and its associated reference base. It's important to note that multiple variants, or alleles, can potentially exist at this same genomic location.
Following the ID column you see REF, ALT and QUAL. These refer to the reference bases, the alternative observed in your samples and the quality the genotyping program gave to this variant call.
The meta-informations section explains the other three fields: FILTER, INFO and FORMAT. You can use FILTER to classify the position the way it's convenient to you. For example: you may want to flag positions with low sequencing coverage, or those positions where you observed balance between reads supporting allele A x B.
After the FORMAT column, you will see columns named with your sample names. They have fields separated by ":" and refer to the fields in the FORMAT column. When you see a field with only a dot it usually represents missing data. For example: if no sequencing reads over the position then the GT field will not have data to show, hence it will have only a dot.
When dealing with tens of thousands of samples the VCF file may not be the best way to proceed. I'm still not very familiar with it but TileDB-VCF seems to be good in this scenario. In fact, the GATK uses GenomicsDB which is built on top of TileDB.
Where to find resources to learn more
To learn how to convert formats, filter, concatenate or merge VCFs, check the official documentation of BCFtools:
If you already are using GATK tools you may want to stay with them. In this case check this other program:
In case you are a developer and need to develop new tools to manipulate VCF files you can find these resources useful:
For Python take a look into the library pysam. Besides the name, it works well on VCF too.
If Go is your language, then check the brentp/vcfgo package.
Probably all major programming languages have libraries to handle this file format. Rust, Nim, R, Java, and others. It's your choice.